

Krankheitsbild
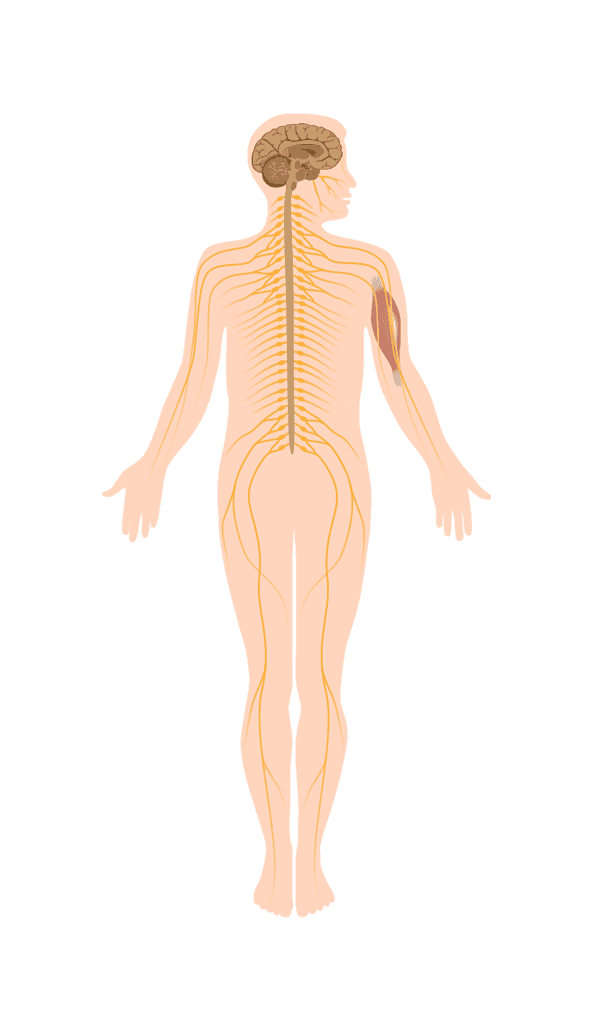
Spinale Muskelatrophie (SMA) ist eine seltene neuromuskuläre Erkrankung. Aufgrund eines „Gendefektes" wird ein bestimmtes Protein, das SMN-Protein, nicht in genügender Menge produziert.
Dadurch erkranken die Motoneuronen (Nervenzellen), die normalerweise Impulse an die Muskeln weiterleiten. Bei SMA sind diese Impulse schwach oder fehlen gänzlich, dadurch entsteht als Folge eine Muskelatrophie (Muskelschwund). SMA hat viele Gesichter und betrifft Menschen unterschiedlich.
Symptome zeigen sich in Einschränkungen von alltäglichen Fähigkeiten wie zu atmen, der Fähigkeit zu essen, sich zu umarmen, etwas zu ergreifen, mit dem Kopf zu nicken, zu sitzen oder zu gehen.
ungefähr eines von 6’000 bis 10’000 Neugeborenen ist betroffen (rare disease, orphan disease)
ungefähr eine von 40 Personen ist Träger des „Gendefektes"
Alle Muskeln des Körpers sind beeinträchtigt, obwohl die so genannten proximalen Muskeln (die dem Rumpf am nächsten sind, z.B. Schulter-, Hüft- und Rückenmuskulatur) oft am schwersten betroffen sind. Die Schwäche in den Beinen ist im Allgemeinen grösser als in den Armen. Es kann auch die Kau- und Schluckmuskulatur betroffen sein.
Die Beteiligung der Atemmuskulatur (die für die Atmung und das Abhusten zuständig ist) kann zu einer erhöhten Anfälligkeit für Lungenentzündungen führen.
Die kognitiven Fähigkeiten, die Hautsensibilität und die Sinneswahrnehmungen sind nicht betroffen.
"Disease-modifying" Medikamente
Heute gibt es sogenannte "Disease-modifying" Medikamente, die den Verlauf der Erkrankung stark verändern.
Kinder, die vor den ersten Symptomen diagnostiziert (z.B. durch Neugeborenen-Screening) und danach medikamentös behandelt werden, gelten als präsymptomatisch. Zur Entwicklung dieser Kinder gibt es noch wenige Daten, aber je früher Kinder behandelt werden können, desto weniger Symptome werden sie im Verlauf ihres Lebens entwickeln.

Bei einer symptomatischen Diagnose wird das breite Spektrum der SMA-Patienten in klinische Typen eingeteilt, die auf dem Alter des Auftretens und der jemals erreichten maximalen motorischen Funktion basieren: Typ 0 (in der Regel tödlich bei der Geburt); Typ 1 (auch: Werdnig Hoffmann; akute infantile SMA; unfähig, unabhängig zu sitzen; Symptome innerhalb der ersten Lebensmonate); Typ 2 (- intermediäre SMA; chronisch infantile SMA; in der Lage, unabhängig zu sitzen, aber nicht zu gehen; Symptome zwischen 7 und 18 Monaten); Typ 3 (Kugelberg-Welander; juvenile SMA; unabhängiges Gehen; Symptome nach 18 Monaten) und Typ 4 (unabhängiges Gehen und Beginn im Erwachsenenalter).
Unterschiede im Krankheitsverlauf unabhängig vom Typ sind häufig, und mit den heutigen medikamentösen Behandlungsmethoden noch zusätzlich von Bedeutung. Daher sind heutige Empfehlungen Betroffene eher nach ihrem aktuellen Funktionsstatus einzuteilen (oder auf einer Kombination von Faktoren):
Non-sitter (diejenigen, die nicht frei sitzen können)
Sitter (die, die frei sitzen können)
Walker (die, die ohne Hilfsmittel frei gehen können)
